by Maria Troppenz & Santiago Rigamonti for exciting oxygen
Purpose: In this tutorial you will learn how to calculate electronic transport coefficients with exciting. The coefficients are obtained by solving the linearized Boltzmann equation in the constant relaxation time approximation. They are calculated for bulk silicon.
|
Table of Contents
|
0. Define relevant environment variables
Read the following paragraphs before starting with the rest of this tutorial!
Before starting, be sure that relevant environment variables are already defined as specified in How to set environment variables for tutorials scripts. The following script is used in this tutorial:
- PLOT-files.py: Python visualization tool for multiple files with the same name in different directories (for more details, see The python script "PLOT-files.py").
From now on the symbol $ will indicate the shell prompt.
Important note: All input parameters are in atomic units!
1. Theoretical background
2. Transport coefficients of Bulk Silicon
To start the tutorial, create a new, empty directory named BE-Si and move into it.
$ mkdir BE-Si
$ cd BE-Si
i) Preparation of the calculation
Save the following lines as input.xml:
<input> <title>Bulk Silicon</title> <structure speciespath="$EXCITINGROOT/species/"> <crystal scale="10.26"> <basevect>0.0 0.5 0.5</basevect> <basevect>0.5 0.0 0.5</basevect> <basevect>0.5 0.5 0.0</basevect> </crystal> <species speciesfile="Si.xml"> <atom coord="0.00 0.00 0.00"></atom> <atom coord="0.25 0.25 0.25"></atom> </species> </structure> <groundstate do="fromscratch" xctype="GGA_PBE_SOL" ngridk="4 4 4" rgkmax="8.0" gmaxvr="14.0" maxscl="100"> </groundstate> </input>
Set up $EXCITINGROOT to the correct root directory in speciespath and execute exciting_smp with the commands
$ SETUP-excitingroot.sh
$ time exciting_smp
To gain a better understanding of the later calculated transport coefficients of silicon, you may want to look at the band structure. For this, please expand the link below and follow the instructions.
ii) Calculation of the transport coefficients at room temperature
In the following, we calculate the transport coefficients with
respect to the chemical potential at a constant temperature (here 300K).
The equations of the transport coefficients (see Section 1) contain the derivative of the Fermi distribution function
To obtain eigenenergies for a fine k-grid, we perform a single self-consistency loop on top of the previous ground-state calculation by changing in input.xml
- the attribute do = "fromfile";
- the attribute ngridk = "40 40 40";
- the attribute maxscl = "1";
in the element groundstate (for more details see Input Reference).
The element groundstate in input.xml should now look like this:
... <groundstate do="fromfile" xctype="GGA_PBE_SOL" ngridk="40 40 40" rgkmax="8.0" gmaxvr="14.0" maxscl="1"> </groundstate> ...
In order to calculate the transport coefficients, delete in your input.xml the "old" properties block that you used for calculating the electronic band-structure:
<properties> <bandstructure> ... </bandstructure> </properties>
and substitute it with
<properties> <momentummatrix/> <boltzequ energyReference="efermi" transportDfRange="-0.5 0.5" transportDfSpacing="0.001" temperatureRange="300 300" temperatureSpacing="1" chemicalPotentialRange="-0.05 0.05" chemicalPotentialSpacing="0.0001" evOutputEnergies="true" siOutputUnits="true"> <etCoeffComponents>1 1</etCoeffComponents> <etCoeffComponents>2 2</etCoeffComponents> <etCoeffComponents>3 3</etCoeffComponents> </boltzequ> </properties>
With inserting the element momentummatrix, the momentum matrix elements are calculated. Inside the element boltzequ, you define the attributes needed for the calculation of the transport coefficients.
An explanation of the attributes can be found by expanding the link below or in Input Reference.
By executing exciting_smp
$ time exciting_smp
the Seebeck coefficient
Please note:
Now, the transport coefficients are calculated and you can plot them by using PLOT-files.py. A detailed description of this plot script can be found in The Python Script PLOT-files.py.
The Seebeck coefficient at the temperature of 300K can be visualized with the following commands:
$ cp SEEBECK_11.OUT S11
$ cp SEEBECK_22.OUT S22
$ cp SEEBECK_33.OUT S33
$ PLOT-files.py -f S11 S22 S33 -cx 2 -cy 3 -ll '$S_{xx}$' '$S_{yy}$' '$S_{zz}$' -x -1 1 -lx '$\mu$ [eV]' -ly 'S [$\mu$V/K]' -ys 1e6 -rp
The output is saved in PLOT.png and PLOT.eps, and looks as follows:

Assuming a relaxation time
$ cp ELECTCOND_11.OUT E11
$ cp ELECTCOND_22.OUT E22
$ cp ELECTCOND_33.OUT E33
$ PLOT-files.py -f E11 E22 E33 -cx 2 -cy 3 -ll '$\sigma_{xx}$' '$\sigma_{yy}$' '$\sigma_{zz}$' -x -0.5 0.5 -y 0 9000 -lx '$\mu$ [eV]' -ly '$\sigma$ [S/cm]' -ys 1e-16 -lp 'upper center' -rp -mty 5
Output:

And the corresponding power factor looks as:
$ paste ELECTCOND_11.OUT SEEBECK_11.OUT | awk '{ printf $1 "\t" $2 "\t" $3*$7*$7 "\n" }' > PF11
$ paste ELECTCOND_22.OUT SEEBECK_22.OUT | awk '{ printf $1 "\t" $2 "\t" $3*$7*$7 "\n" }' > PF22
$ paste ELECTCOND_33.OUT SEEBECK_33.OUT | awk '{ printf $1 "\t" $2 "\t" $3*$7*$7 "\n" }' > PF33
$ PLOT-files.py -f PF11 PF22 PF33 -cx 2 -cy 3 -ll '${\sigma S^2}_{xx}$' '${\sigma S^2}_{yy}$' '${\sigma S^2}_{zz}$' -x -0.5 0.5 -y 0 20 -lx '$\mu$ [eV]' -ly '$\sigma S^2$ [$\mu W\,cm^{-1}\,K^{-2}$]' -ys 1e-10 -lp 'upper center' -rp
Output:

iii) Calculation of the transport coefficients at different doping concentrations
In the following, we look at the temperature dependence of transport coefficients at a constant chemical potential.
We can use the fine k-grid obtained in part ii) and skip the ground-state calculation by changing in input.xml
- The attribute do to "skip" in the element groundstate
- The momentum matrix elements does not need to be calculated again and, e.g., can be removed or commented.
The element properties of your input.xml can be changed to
<properties> <!--momentummatrix/--> <boltzequ energyReference="efermi" transportDfRange="-0.5 0.5" transportDfSpacing="0.001" temperatureRange="300 1200" temperatureSpacing="10" chemicalPotentialRange="-0.005 0.005" chemicalPotentialSpacing="0.005" evOutputEnergies="true" siOutputUnits="true"> <etCoeffComponents>1 1</etCoeffComponents> </boltzequ> </properties>
The transport coefficients are calculated from 300 to 1200 K and for the chemical potentials
With the modified input.xml, we can rerun exciting_smp
$ time exciting_smp &
The Seebeck coefficient for
$ grep ' -0.136' SEEBECK_11.OUT > S11M
$ grep ' 0.136' SEEBECK_11.OUT > S11P
$ PLOT-files.py -f S11P S11M -cy 3 -ll '$S_{xx}$@$\mu$= 0.136eV' '$S_{xx}$@$\mu$=-0.136eV' -x 300 1200 -lx 'T [K]' -ly 'S [$\mu$V/K]' -ys 1e6 -lp 5 -rp
Output:

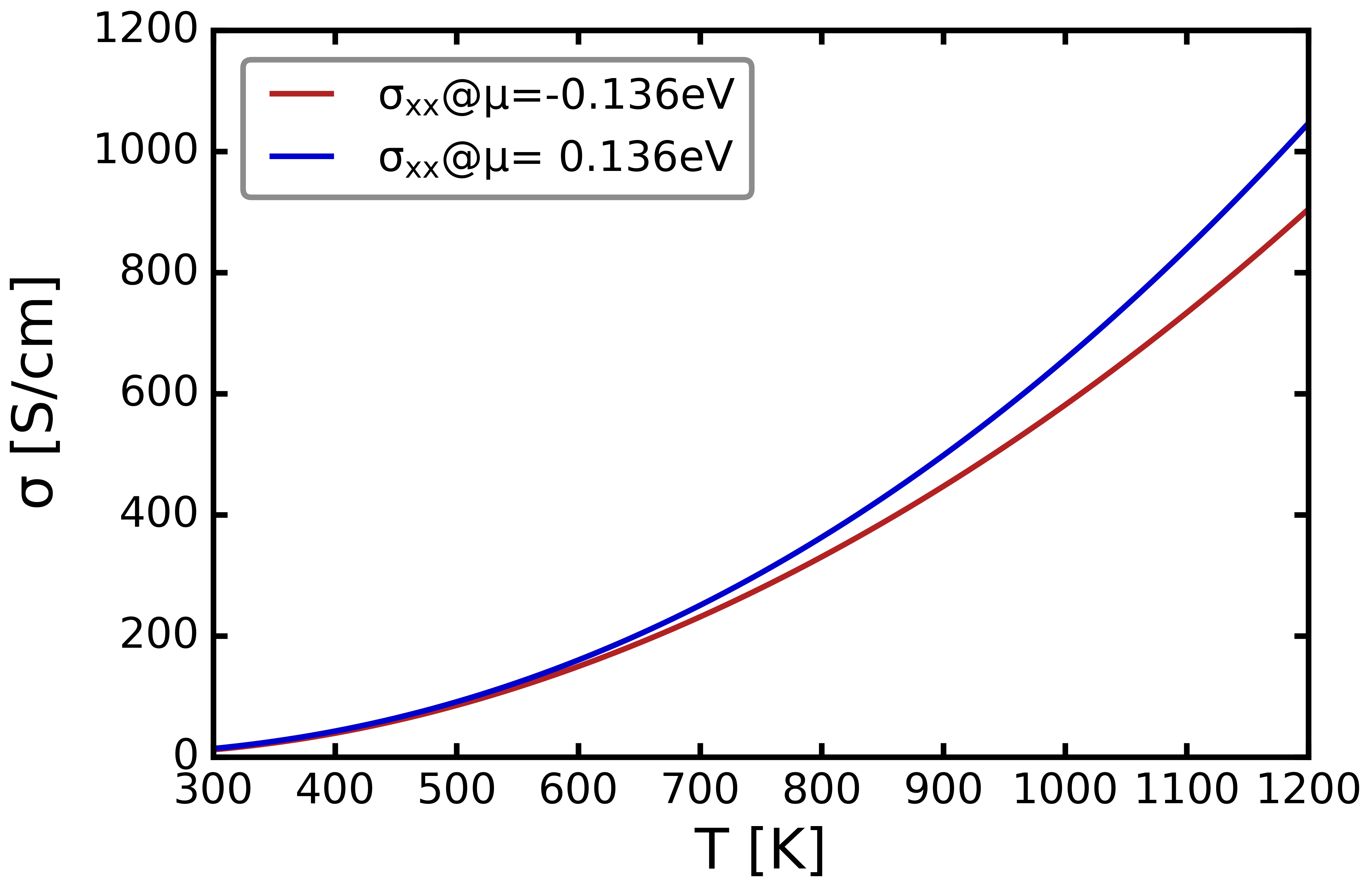
Again, using
$ grep ' -0.136' ELECTCOND_11.OUT > E11M
$ grep ' 0.136' ELECTCOND_11.OUT > E11P
$ PLOT-files.py -f E11P E11M -cy 3 -ll '$\sigma_{xx}$@$\mu$= 0.136eV' '$\sigma_{xx}$@$\mu$=-0.136eV' -x 300 1200 -lx 'T [K]' -ly '$\sigma$ [S/cm]' -ys 1e-16 -rp
Output:
And the corresponding power factor is
$ paste ELECTCOND_11.OUT SEEBECK_11.OUT | grep ' -0.136' | awk '{ printf $1 "\t" $2 "\t" $3*$7*$7 "\n" }' > PF11M
$ paste ELECTCOND_11.OUT SEEBECK_11.OUT | grep ' 0.136' | awk '{ printf $1 "\t" $2 "\t" $3*$7*$7 "\n" }' > PF11P
$ PLOT-files.py -f PF11P PF11M -cy 3 -ll '${\sigma S^2}_{xx}$@$\mu$= 0.136eV' '${\sigma S^2}_{xx}$@$\mu$=-0.136eV' -x 300 1200 -lx 'T [K]' -ly '$\sigma S^2$ [$\mu W\,cm^{-1}\,K^{-2}$]' -ys 1e-10 -rp
Output:










0 Comments